Angiotensin 1/2 (1-6)Vasoconstrictor CAS# 47896-63-9 |
")
- Celastrol
Catalog No.:BCN5986
CAS No.:34157-83-0
- BMS-345541
Catalog No.:BCC1423
CAS No.:547757-23-3
- Bay 65-1942 free base
Catalog No.:BCC1408
CAS No.:600734-02-9
- Bay 65-1942 HCl salt
Catalog No.:BCC1409
CAS No.:600734-06-3
- Bay 65-1942 R form
Catalog No.:BCC1410
CAS No.:758683-21-5
Quality Control & MSDS
3D structure
Package In Stock

Number of papers citing our products

| Cas No. | 47896-63-9 | SDF | Download SDF |
| PubChem ID | 9832262 | Appearance | Powder |
| Formula | C36H55N11O10 | M.Wt | 801.89 |
| Type of Compound | N/A | Storage | Desiccate at -20°C |
| Solubility | >80.2mg/ml in DMSO | ||
| Sequence | H2N-Asp-Arg-Val-Tyr-Ile-His-OH | ||
| Chemical Name | (3S)-3-amino-4-[[(2S)-1-[[(2S)-1-[[(2S)-1-[[(2S,3S)-1-[[(1S)-1-carboxy-2-(1H-imidazol-5-yl)ethyl]amino]-3-methyl-1-oxopentan-2-yl]amino]-3-(4-hydroxyphenyl)-1-oxopropan-2-yl]amino]-3-methyl-1-oxobutan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-4-oxobutanoic acid | ||
| SMILES | CCC(C)C(C(=O)NC(CC1=CN=CN1)C(=O)O)NC(=O)C(CC2=CC=C(C=C2)O)NC(=O)C(C(C)C)NC(=O)C(CCCN=C(N)N)NC(=O)C(CC(=O)O)N | ||
| Standard InChIKey | SYDDLSICWJNDAM-GKUXVWPZSA-N | ||
| Standard InChI | InChI=1S/C36H55N11O10/c1-5-19(4)29(34(55)45-26(35(56)57)14-21-16-40-17-42-21)47-32(53)25(13-20-8-10-22(48)11-9-20)44-33(54)28(18(2)3)46-31(52)24(7-6-12-41-36(38)39)43-30(51)23(37)15-27(49)50/h8-11,16-19,23-26,28-29,48H,5-7,12-15,37H2,1-4H3,(H,40,42)(H,43,51)(H,44,54)(H,45,55)(H,46,52)(H,47,53)(H,49,50)(H,56,57)(H4,38,39,41)/t19-,23-,24-,25-,26-,28-,29-/m0/s1 | ||
| General tips | For obtaining a higher solubility , please warm the tube at 37 ℃ and shake it in the ultrasonic bath for a while.Stock solution can be stored below -20℃ for several months. We recommend that you prepare and use the solution on the same day. However, if the test schedule requires, the stock solutions can be prepared in advance, and the stock solution must be sealed and stored below -20℃. In general, the stock solution can be kept for several months. Before use, we recommend that you leave the vial at room temperature for at least an hour before opening it. |
||
| About Packaging | 1. The packaging of the product may be reversed during transportation, cause the high purity compounds to adhere to the neck or cap of the vial.Take the vail out of its packaging and shake gently until the compounds fall to the bottom of the vial. 2. For liquid products, please centrifuge at 500xg to gather the liquid to the bottom of the vial. 3. Try to avoid loss or contamination during the experiment. |
||
| Shipping Condition | Packaging according to customer requirements(5mg, 10mg, 20mg and more). Ship via FedEx, DHL, UPS, EMS or other couriers with RT, or blue ice upon request. | ||

Angiotensin 1/2 (1-6) Dilution Calculator
Angiotensin 1/2 (1-6) Molarity Calculator

Calcutta University

University of Minnesota

University of Maryland School of Medicine

University of Illinois at Chicago

The Ohio State University

University of Zurich

Harvard University

Colorado State University

Auburn University

Yale University

Worcester Polytechnic Institute

Washington State University

Stanford University

University of Leipzig

Universidade da Beira Interior

The Institute of Cancer Research

Heidelberg University

University of Amsterdam

University of Auckland

TsingHua University

The University of Michigan

Miami University

DRURY University

Jilin University

Fudan University

Wuhan University

Sun Yat-sen University

Universite de Paris

Deemed University

Auckland University

The University of Tokyo

Korea University
Angiotensin I/II (1-6) is a peptide (ASP-ARG-VAL-TYR-ILE-HIS) that contains the amino acids 1-6 and is converted from Angiotensin I/II peptide.
Angiotensin I is formed by the action of renin on angiotensinogen, an α-2-globulin with 12 amino acids. Angiotensinogen is produced constitutively and released into the circulation mainly by the liver. Renin cleaves the peptide bond between the leucine (Leu) and valine (Val) residues on angiotensinogen, creating the ten-amino acid peptide angiotensin I. Angiotensin I is converted to angiotensin II (AII) through removal of two C-terminal residues by angiotensin-converting enzyme (ACE), primarily by ACE within the lung.
Angiotensin is a peptide hormone that causes vasoconstriction and a subsequent increase in blood pressure. Angiotensin also stimulates the release of aldosterone, which promotes sodium retention in the distal nephron, thereby increasing blood pressure.
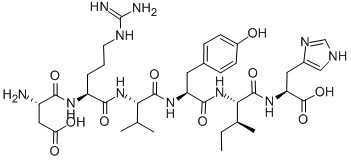
Figure1 Formula of Angiotensin I/II (1-6)
Ref:
1. Basso N, Terragno NA (December 2001). "History about the discovery of the renin-angiotensin system". Hypertension 38 (6): 1246–9.
2. Richard A. Preston. et. (1998). “Age-Race Subgroup Compared With Renin Profile as Predictors of Blood Pressure Response to Antihypertensive Therapy”. JAMA. 1998;280(13):1168-1172.
3. Williams GH, Dluhy RG (2008). "Chapter 336: Disorders of the Adrenal Cortex". In Loscalzo J, Fauci AS, Braunwald E, Kasper DL, Hauser SL, Longo DL. Harrison's principles of internal medicine. McGraw-Hill Medical.
- Zopfiellamide A
Catalog No.:BCN1865
CAS No.:478945-64-1
- Kisspeptin 10 (rat)
Catalog No.:BCC6132
CAS No.:478507-53-8
- CP 339818 hydrochloride
Catalog No.:BCC7048
CAS No.:478341-55-8
- ISO-1
Catalog No.:BCC5427
CAS No.:478336-92-4
- 4-Hydroxymethylphenol 1-O-rhamnoside
Catalog No.:BCN7750
CAS No.:478314-67-9
- Gabapentin enacarbil
Catalog No.:BCC4239
CAS No.:478296-72-9
- R-1479
Catalog No.:BCC1878
CAS No.:478182-28-4
- Isoerysenegalensein E
Catalog No.:BCN3978
CAS No.:478158-77-9
- PHA 543613 hydrochloride
Catalog No.:BCC5972
CAS No.:478149-53-0
- Pseudococaine
Catalog No.:BCN1902
CAS No.:478-73-9
- Berbamine
Catalog No.:BCN5543
CAS No.:478-61-5
- Rhein
Catalog No.:BCN5947
CAS No.:478-43-3
- Calcium-Sensing Receptor Antagonists I
Catalog No.:BCC1448
CAS No.:478963-79-0
- Coumestrol
Catalog No.:BCN3949
CAS No.:479-13-0
- Dyphylline
Catalog No.:BCC2297
CAS No.:479-18-5
- Atranorin
Catalog No.:BCN5544
CAS No.:479-20-9
- Cotoin
Catalog No.:BCN5545
CAS No.:479-21-0
- Indirubin
Catalog No.:BCN2385
CAS No.:479-41-4
- Canthin-6-one
Catalog No.:BCN5546
CAS No.:479-43-6
- Artemetin
Catalog No.:BCN5547
CAS No.:479-90-3
- Vitexicarpin
Catalog No.:BCN5020
CAS No.:479-91-4
- Aucubin
Catalog No.:BCN5355
CAS No.:479-98-1
- [Orn8]-Urotensin II
Catalog No.:BCC5793
CAS No.:479065-85-5
- MMPIP hydrochloride
Catalog No.:BCC7528
CAS No.:479077-02-6
Deletion of angiotensin-converting enzyme 2 exacerbates renal inflammation and injury in apolipoprotein E-deficient mice through modulation of the nephrin and TNF-alpha-TNFRSF1A signaling.[Pubmed:26245758]
J Transl Med. 2015 Aug 6;13:255.
BACKGROUND: The renin-angiotensin system (RAS) has been implicated in atherosclerotic lesions and progression to chronic kidney diseases. We examined regulatory roles of angiotensin-converting enzyme 2 (ACE2) in the apolipoprotein E (ApoE) knockout (KO) kidneys. METHODS: The 3-month-old wild-type, ApoEKO, ACE2KO and ApoE/ACE2 double-KO (DKO) mice in a C57BL/6 background were used. The ApoEKO mice were randomized to daily deliver either Ang II (1.5 mg/kg) and/or human recombinant ACE2 (rhACE2; 2 mg/kg) for 2 weeks. We examined changes in pro-inflammatory cytokines, renal ultrastructure, and pathological signaling in mouse kidneys. RESULTS: Downregulation of ACE2 and nephrin levels was observed in ApoEKO kidneys. Genetic ACE2 deletion resulted in modest elevations in systolic blood pressure levels and Ang II type 1 receptor expression and reduced nephrin expression in kidneys of the ApoE/ACE2 DKO mice with a decrease in renal Ang-(1-7) levels. These changes were linked with marked increases in renal superoxide generation, NADPH oxidase (NOX) 4 and proinflammatory factors levels, including interleukin (IL)-1beta, IL-6, IL-17A, RANTES, ICAM-1, Tumor necrosis factor-alpha (TNF-alpha) and TNFRSF1A. Renal dysfunction and ultrastructure injury were aggravated in the ApoE/ACE2 DKO mice and Ang II-infused ApoEKO mice with increased plasma levels of creatinine, blood urea nitrogen and enhanced levels of Ang II in plasma and kidneys. The Ang II-mediated reductions of renal ACE2 and nephrin levels in ApoEKO mice were remarkably rescued by rhACE2 supplementation, along with augmentation of renal Ang-(1-7) levels. More importantly, rhACE2 treatment significantly reversed Ang II-induced renal inflammation, superoxide generation, kidney dysfunction and adverse renal injury in ApoEKO mice with suppression of the NOX4 and TNF-alpha-TNFRSF1A signaling. However, rhACE2 had no effect on renal NOX2 and TNFRSF1B expression and circulating lipid levels. CONCLUSIONS: ACE2 deficiency exacerbates kidney inflammation, oxidative stress and adverse renal injury in the ApoE-mutant mice through modulation of the nephrin, NOX4 and TNF-alpha-TNFRSF1A signaling. While rhACE2 supplementation alleviates inflammation, renal dysfunction and glomerulus injury in the ApoE-mutant mice associated with upregulations of Ang-(1-7) levels and nephrin expression and suppression of the TNF-alpha-TNFRSF1A signaling. Strategies aimed at enhancing the ACE2/Ang-(1-7) actions may have important therapeutic potential for atherosclerotic renal injury and kidney diseases.
The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway.[Pubmed:25089563]
Antioxid Redox Signal. 2015 Jan 20;22(3):241-58.
UNLABELLED: Reactive oxygen species (ROS) generated by NADPH oxidase-4 (NOX4) have been shown to initiate lung fibrosis. The migration of lung fibroblasts to the injured area is a crucial early step in lung fibrosis. The angiotensin-converting enzyme 2 (ACE2)/angiotensin (1-7) [Ang(1-7)]/Mas axis, which counteracts the ACE/angiotensin II (AngII)/angiotensin II type 1 receptor (AT1R) axis, has been shown to attenuate pulmonary fibrosis. Nevertheless, the exact molecular mechanism remains unclear. AIMS: To investigate the different effects of the two axes of the renin-angiotensin system (RAS) on lung fibroblast migration and extracellular matrix accumulation by regulating the NOX4-derived ROS-mediated RhoA/Rho kinase (Rock) pathway. RESULTS: In vitro, AngII significantly increased the NOX4 level and ROS production in lung fibroblasts, which stimulated cell migration and alpha-collagen I synthesis through the RhoA/Rock pathway. These effects were attenuated by N-acetylcysteine (NAC), diphenylene iodonium, and NOX4 RNA interference. Moreover, Ang(1-7) and lentivirus-mediated ACE2 (lentiACE2) suppressed AngII-induced migration and alpha-collagen I synthesis by inhibiting the NOX4-derived ROS-mediated RhoA/Rock pathway. However, Ang(1-7) alone exerted analogous effects on AngII. In vivo, constant infusion with Ang(1-7) or intratracheal instillation with lenti-ACE2 shifted the RAS balance toward the ACE2/Ang(1-7)/Mas axis, alleviated bleomycin-induced lung fibrosis, and inhibited the RhoA/Rock pathway by reducing NOX4-derived ROS. INNOVATION: This study suggests that the ACE2/Ang(1-7)/Mas axis may be targeted by novel pharmacological antioxidant strategies to treat lung fibrosis induced by AngII-mediated ROS. CONCLUSION: The ACE2/Ang(1-7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rock pathway.
Transgenic Mice Overexpressing Human Angiotensin I Receptor Gene Are Susceptible to Stroke Injury.[Pubmed:25652270]
Mol Neurobiol. 2016 Apr;53(3):1533-1539.
Hypertension is one of the co-morbid conditions for stroke and profoundly increases its incidence. Angiotensin II (AngII) is shown to be at the center stage in driving the renin angiotensin system via activation of angiotensin 1 receptor (AT1R). This makes the AT1R gene one of the candidates whose differential regulation leads to the predisposition to disorders associated with hypertension. A haplotype block of four SNPs is represented primarily by haplotype-I, or Hap-I (TTAA), and haplotype-II, or Hap-II (AGCG), in the promoter of human AT1R (hAT1R) gene. To better understand the physiological role of these haplotypes, transgenic (TG) mice containing Hap-I and Hap-II of the hAT1R gene in a 166-kb bacterial artificial chromosome (BAC) were generated. Mice received injection of endothelin-1 (1 mg/ml) directly in to the striatum and were evaluated for neurologic deficit scores and sacrificed for analysis of infarct volume and mRNA levels of various proteins. Mice containing Hap-I suffered from significantly higher neurological deficits and larger brain infarcts than Hap II. Similarly, the molecular analysis of oxidant and inflammatory markers in brains of mice showed a significant increase (p < 0.05) in NOX-1 (2.3-fold), CRP (4.3-fold), and IL6 (1.9-fold) and a corresponding reduced expression of antioxidants SOD (60%) and HO1 (55%) in Hap-I mice as compared to Hap-II mice. These results suggest that increased expression of hAT1R rendered Hap-I TG mice susceptible to stroke-related pathology, possibly due to increased level of brain inflammatory and oxidative stress markers and a suppressed antioxidant defense system.
Angiotensin-(1-7) Improves Liver Fibrosis by Regulating the NLRP3 Inflammasome via Redox Balance Modulation.[Pubmed:26728324]
Antioxid Redox Signal. 2016 May 10;24(14):795-812.
AIMS: Angiotensin II (Ang II) aggravates hepatic fibrosis by inducing NADPH oxidase (NOX)-dependent oxidative stress. Angiotensin-(1-7) [Ang-(1-7)], which counter-regulates Ang II, has been evidenced to protect against hepatic fibrosis. The NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome, being activated by reactive oxygen species (ROS), is identified as a novel mechanism of liver fibrosis. However, whether the NLRP3 inflammasome involves in regulation of Ang II-induced hepatic fibrosis remains unclear. This study investigates the different effects of the Ang II and Ang-(1-7) on collagen synthesis by regulating the NLRP3 inflammasome/Smad pathway via redox balance modulation. RESULTS: In vivo, Ang-(1-7) improved bile duct ligation-induced hepatic fibrosis, reduced H2O2 content, protein levels of NOX4, and the NLRP3 inflammasome, whereas it increased glutathione (GSH) and nuclear erythroid 2-related factor 2 (Nrf2) antioxidant response element (ARE). In vitro, Ang II treatment elevated NOX4 protein expression and ROS production in hepatic stellate cells (HSCs), whereas it inhibited GSH and Nrf2-ARE, resulting in the activation of the NLRP3 inflammasome in the mitochondria of HSCs. NLRP3 depletion inhibited Ang II-induced collagen synthesis. Furthermore, Ang II increased NLRP3 and pro-IL-1beta levels by activating the Toll-like receptor 4 (TLR4)/MyD88/NF-kappaB pathway. Treatment with antioxidants, NOX4 small interference RNA (siRNA), or Nrf2 activator inhibited Ang II-induced NLRP3 inflammasome activation and collagen synthesis. In contrast, the action of Ang-(1-7) opposed the effects of Ang II. INNOVATION AND CONCLUSIONS: Ang-(1-7) improved liver fibrosis by regulating NLRP3 inflammasome activation induced by Ang II-mediated ROS via redox balance modulation. Antioxid. Redox Signal. 24, 795-812.
MAS receptors mediate vasoprotective and atheroprotective effects of candesartan upon the recovery of vascular angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axis functionality.[Pubmed:26144375]
Eur J Pharmacol. 2015 Oct 5;764:173-88.
AT1 antagonists effectively prevent atherosclerosis since AT1 upregulation and angiotensin II-induced proinflammatory actions are critical to atherogenesis. Despite the classic mechanisms underlying the vasoprotective and atheroprotective actions of AT1 antagonists, the cross-talk between angiotensin-converting enzyme-angiotensin II-AT1 and angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axes suggests other mechanisms beyond AT1 blockage in such effects. For instance, angiotensin-converting enzyme 2 activity is inhibited by reactive oxygen species derived from AT1-mediated proinflammatory signaling. Since angiotensin-(1-7) promotes antiatherogenic effects, we hypothesized that the vasoprotective and atheroprotective effects of AT1 antagonists could result from their inhibitory effects on the AT1-mediated negative modulation of vascular angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axis functionality. Interestingly, our results showed that early atherosclerosis triggered in thoracic aorta from high cholesterol fed-Apolipoprotein E-deficient mice impairs angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axis functionality by a proinflammatory-redox AT1-mediated pathway. In such mechanism, AT1 activation leads to the aortic release of tumor necrosis factor-alpha, which stimulates NAD(P)H oxidase/Nox1-driven generation of superoxide and hydrogen peroxide. While hydrogen peroxide inhibits angiotensin-converting enzyme 2 activity, superoxide impairs MAS functionality. Candesartan treatment restored the functionality of angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axis by inhibiting the proinflammatory-redox AT1-mediated mechanism. Candesartan also promoted vasoprotective and atheroprotective effects that were mediated by MAS since A779 (MAS antagonist) co-treatment inhibited them. The role of MAS receptors as the final mediators of the vasoprotective and atheroprotective effects of candesartan was supported by the vascular actions of angiotensin-(1-7) upon the recovery of the functionality of vascular angiotensin-converting enzyme 2-angiotensin-(1-7)-MAS axis.
